{kind=link}
Лекторий: АКНЕ. Новый образовательный проект для врачей дерматовенерологов и косметологов
вт., 16/11/2021 - 20:39 — главный редактор 
Современная терапия осложнений пигментной ксеродермы
вт., 16/11/2021 - 17:32 — mordovtsevaИзвестно, что частыми осложнениями пигментной ксеродермы являются злокачественные новообразования на коже открытых участков тела. Эти новообразования, как правило, множественные.
Новые кожные опухоли образуются с высокой скоростью и трудно поддаются лечению.
Прежде всего, необходимо помнить в важности ранней диагностики заболевания, строгого противопроказания инсоляции и других источников ультрафиолетового излучения. На улице нельзя находиться без соответствующей защиты, даже в пасмурные дни.
При малейшем подозрении на пигментную ксеродерму необходимо избегать солнца и других источников уф излучения до подтверждения или опровержения диагноза. Обязателен прием витамина Д.
Для профилактики злокачественных новообразований рекомендованы изотретиноин и ацитретин. Однако, учитывая их токсичность, эти препараты следует назначать только больным, у которых быстро развиваются множественные опухоли кожи.
О кутавирусе
вт., 16/11/2021 - 15:59 — Юрий_Боровиков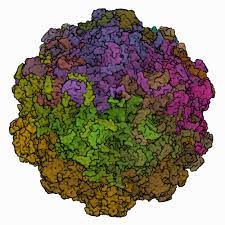 Кутавирус (Cutavirus) – новый член семейства парвовирусов, первые сообщения о котором появились в 2016 году.
Кутавирус (Cutavirus) – новый член семейства парвовирусов, первые сообщения о котором появились в 2016 году.
О нем пишут разное.
Ученые из Финляндии (doi.org/10.1093/cid/ciy806) сообщают об обнаружении ДНК кутавируса в биоптатах кожи у 4-х из 25 пациентов с Т-клеточной лимфомой кожи и у 4-х из 136 обследуемых, перенесших трансплантацию внутренних органов. Вместе с этим, в коже здоровых людей кутавирус найден не был.
Эспрессо для волос
сб., 13/11/2021 - 21:57 — Мордовцева Валерия ВладимировнаУже в течение ряда лет кофеин входит в состав некоторых лекарственных препаратов (в частности, анальгетиков), а также косметических средств, например, шампуня «против выпадения волос». Что же служит основанием для применения кофеина в трихологии?
Ответ на этот вопрос дается в статье, ссылка на которую в конце данного материала. Фактически это квинтэссенция результатов изучения воздействия кофеина на волосяной фолликул, имеющихся к настоящему времени:
При сочетании наружного тестостерона в концентрациях 5 нгр - 5 мкг/мл и кофеина в концентрациях 10 – 1500 мкг/мл было обнаружено, что тестостерон подавлял рост фолликулов, а на фоне применения кофеина в дозе 10 и 50 мкг/мл этот эффект нивелировался.
При отсутствии тестостерона данные дозы кофеина стимулировали рост волос, а также пролиферацию кератиноцитов в области дермального сосочка. При этом более высокие дозы кофеина (100, 500 и 1500 мкг на мл) оказывали ингибирующий эффект.
Авторы объясняют это тем, что высокие дозы кофеина вызывают гиперстимуляцию метаболизма волосяного фолликула, что сопровождается истощением энергетических ресурсов и пролиферативной способности и, как следствие, отсутствием удлинения волосяного стержня.
В низких концентрациях кофеин способствует удлинению анагена и противодействует экспрессии протеина TGF-beta2 в волосяных фолликулах у мужчин с андрогенетической алопецией.
У здоровых женщин наружный тестостерон не оказывает никакого влияния на TGF-beta2, однако, при сочетании с кофеином, отмечается значительное снижение экспрессии этого белка.
Как у мужчин, так и у женщин, кофеин усиливает экспрессию белка IGF-1, что приводит к увеличению продолжительности анагеновой фазы. (Известно, что для поддержания анагеновой фазы необходимы высокая экспрессия IGF-1 и низкая – TGF-beta2).
Ушел из жизни академик Николай Сергеевич Потекаев (1924-2021)
пн., 08/11/2021 - 19:20 — главный редактор Российская дерматовенерологическая школа понесла тяжелую утрату. На 98-м году ушел из жизни выдающийся ученый, блестящий клиницист и педагог, член-корреспондент РАН, Заслуженный профессор ПМГМУ имени И.М. Сеченова, лауреат Премии Совета Министров СССР Потекаев Николай Сергеевич. Профессор Потекаев отдал своей профессии почти 70 лет, до последних дней занимаясь научной работой.
Российская дерматовенерологическая школа понесла тяжелую утрату. На 98-м году ушел из жизни выдающийся ученый, блестящий клиницист и педагог, член-корреспондент РАН, Заслуженный профессор ПМГМУ имени И.М. Сеченова, лауреат Премии Совета Министров СССР Потекаев Николай Сергеевич. Профессор Потекаев отдал своей профессии почти 70 лет, до последних дней занимаясь научной работой.
Николай Сергеевич родился 17 сентября 1924 года в деревне Ново-Самарка Сорочинского района Оренбургской области. В 1942 году после окончания пехотного училища лейтенантом ушел на фронт, командовал взводом противотанковой пехоты, участвовал в сражении на Курской дуге, где получил тяжелое ранение. Награжден Орденом отечественной войны II степени, медалями «За победу над Германией» и «За доблестный труд в Великой отечественной войне».
В 1946 году Н.С. Потекаев поступил на первый курс лечебного факультета I Московского медицинского института им. И.М. Сеченова (ныне Первый Московский государственный медицинский университет имени И.М. Сеченова), который окончил с отличием в 1952 году. Он проработал здесь более 50 лет, пройдя путь от ординатора до заведующего кафедрой. В 1988 году он был избан членом-корреспондентом Академии Медицинских Наук СССР.
Андрогенетическая алопеция и дютастерид
вс., 07/11/2021 - 19:27 — Мордовцева Валерия ВладимировнаСогласно данным литературы, дютастерид, представляющий собой блокатор 5-альфа-редуктазы, в настоящее время разрешен к применению при андрогенетической алопеции только в Японии и Южной Корее, хотя дерматологи и в других странах применяют его «не по показаниям».
Недавние исследования указывают на то, что пероральный дютастерид в 3 раза более эффективно ингибирует 5-альфа-редуктазу 2 типа и является в 100 раз более эффективным в плане ингибирования 5-альфа-редуктазы 1 типа.
С учетом риска развития побочных эффектов системных антиандрогенов альтернативой явилось бы использование их в качестве наружных средств. И в этом случае пальма первенства принадлежала бы дютастериду.
Нант
вт., 02/11/2021 - 16:05 — Юрий_Боровиков Когда-то респектабельный буржуазный Нант был одним из крупнейших мировых центров работорговли. Отсюда к берегам Африки уходили корабли с ситцами, галантерейным товаром, металлическими, стеклянными украшениями и алкоголем.
Когда-то респектабельный буржуазный Нант был одним из крупнейших мировых центров работорговли. Отсюда к берегам Африки уходили корабли с ситцами, галантерейным товаром, металлическими, стеклянными украшениями и алкоголем.
Добравшись до “черного” континента, нантские купцы обменивали свой груз на живой товар и далее направляли свои суда, трюмы которых были забиты рабами, в сторону Америки. В среднем за год работорговцы из Нанта скупали в Африке 8-9 тысяч рабов, из которых около полутора тысяч погибало в пути.
Решение о создании в Нанте Мемориала отмены рабства далось его жителям нелегко. Все-таки это темная страница в истории города.
Однако, нантцы сумели осознать этот исторический факт, принять на себя ответственность за прошлое и призвать мир противостоять всем современным формам рабства.
Мемориал представляет собой удивительный архитектурный объект. Это часть набережной реки Луары, в покрытие которой хаотично вставлены более 2000 стеклянных табличек с датами и названиями судов экспедиций работорговцев.
Шалфей и волосы
вс., 31/10/2021 - 03:56 — Мордовцева Валерия ВладимировнаРечь пойдет о шалфее и о его возможном использовании в трихологии. Guang-Ri Jin и соавт., изучили влияние экстракта шалфея на клетки волосяного фолликула.
Растение целиком измельчали до состояния порошка, затем добавляли 99,9% метиловый спирт. Разными концентрациями полученного экстракта обрабатывали культуру клеток волосяного фолликула человека.
Затем в ходе наблюдения оценивали пролиферацию и миграцию клеток, а также различные факторы, связанные с циклом развития волосяного фолликула и ростом волос.
Кроме того, экстракт применяли местно у мышей в течение 21 дня.
Пигментная ксеродерма и онкогематология
сб., 30/10/2021 - 19:11 — mordovtsevaКак известно, при пигментной ксеродерме, редком генодерматозе с аутосомно-рецессивным (в большинстсе случаев) типом наследования, имеет место дефект репарации ДНК, поврежденной солнечной радиацией.
В основе развития пигментной ксеродермы лежат мутации в генах XPA, ERCC3/XPB, XPC, ERCC2/XPD, DDB2/XPE, ERCC4/XPF/FANQ и ERCC5/XPG или же в гене POLH/HPV, который кодирует обратную транскриптазу.
При большинстве синдромов, обусловленных нарушениями репарации ДНК, клинические дерматологические признаки и злокачественные новообразования кожи развиваются уже в детском возрасте, а у 25% больных наблюдаются прогрессирующие дегенеративные неврологические изменения.
При пигментной ксеродерме имеет место выраженное повышенние частоты развития злокачественных новообразований кожи, таких как базальноклеточный и плоскоклеточный рак, ангио- и фибросаркома, меланома (риск, в 10 000 превышающий таковой в обычной популяции), а также преждевременное старение, нейросенсорная глухота и ускоренная утрата нейронов.
Клетки мозга и рост волос
сб., 30/10/2021 - 12:33 — Мордовцева Валерия ВладимировнаВолосяной фолликул, «живущий» во всех слоях кожи, является динамичным мини-органом. Его образуют 20 типов клеток. Известно, что клетки волосяного сосочка играют важную роль в морфогенезе фолликула: от них зависит поддержание анагеновой фазы, длина волосяного стержня и его форма.
Для успешного лечения выпадения волос необходимо поддержание количества клеток дермального сосочка на определенном уровне и/или ускорение пролиферации этих клеток.
Если бы переход из телогена в анаген был более быстрым, или же переход из анагена в катаген более медленным, то, соответственно, и уменьшение размеров волосяных фолликулов и выпадение волос было бы менее выражено.
| © Дерматология в России, 2007-2025
|
О сайте | Правовая информация | |
| Материалы сайта предназначены только для врачей. По вопросам, касающимся Вашего здоровья, обратитесь к специалисту. 18+ | |||